You have no items in your shopping cart.
Hallmarks of Cancer
Cancer Background
Despite the many recent advances in our understanding and treatment of the disease, cancer is still among the leading causes of death worldwide. In 2018, there were 18.1 million new cases and 9.5 million cancer-related deaths worldwide, with cancer being the second leading cause of death in the United States of America.
- About one-third of all people in the US will develop cancer during their lifetimes.
- By 2040, the number of new cancer cases per year is expected to rise to 29.5 million and cancer-related deaths to 16.4 million.
Generally, cancer rates are highest in countries with the longest life expectancy, education level, and living standards, such as the US, Canada, and the UK. However, for some cancer types, such as cervical cancer, the reverse is observed.
Cancer: Causes and Treatments
Cancer is caused by a wide range of environmental factors, leading to DNA damage, hereditary and in utero mutations.
- Environmental factors, such as smoking, excessive alcohol consumption, and radiation exposure, contribute to an individual's lifetime risk of developing cancer.
- Inherited genetic mutations play a significant role in around 5 -10% of all cancers.
More than 50 mutations in specific genes have been linked to hereditary cancer syndromes.
Hallmarks of Cancer
The canonical hallmarks of cancer comprise six physical capabilities acquired during the multistep development of human tumors, built on a foundation of genomic instability and inflammation. Research and increased understanding of cancer in the last decade have provided two emerging hallmarks that contribute to establishing the core capabilities-reprogramming of energy metabolism and evading immune destruction. In addition to cancer cells, tumors exhibit another dimension of complexity: they contain a repertoire of recruited, ostensibly normal cells that contribute to the acquisition of hallmark traits by creating the "tumor microenvironment." Recognition of these concepts' widespread applicability increasingly affect the development of new means to treat human cancer.
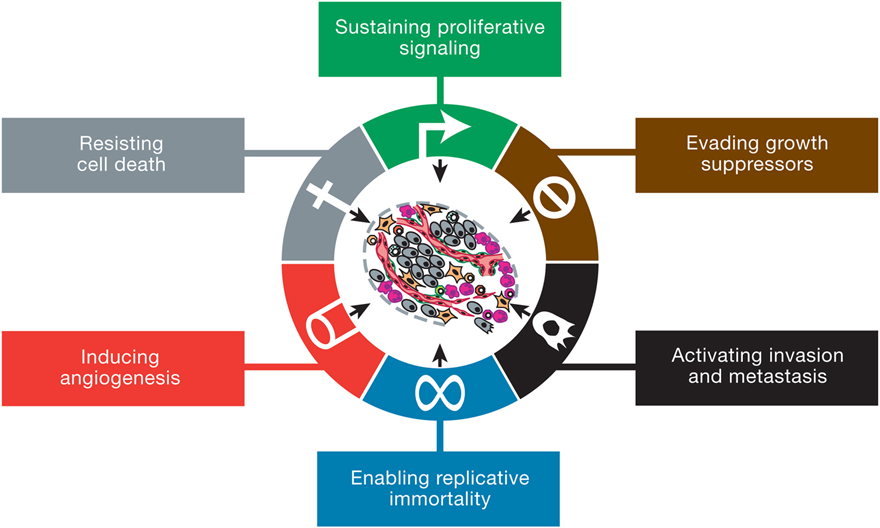
Fig. 1. The Hallmarks of Cancer. Source: Hanahan and Weinberg, 2011
Sustaining Proliferative Signaling
A fundamental trait of cancer cells is their ability to support chronic proliferation. Healthy tissues carefully regulate the cell growth-and-division cycle, ensuring homeostasis and standard tissue architecture and function. In contrast, cancer cells deregulate these signaling pathways to maintain their survival. Growth factors (GF) that bind to cell-surface receptors containing intracellular tyrosine kinase domains are predominantly affected. This leads to the upregulation of intracellular signaling pathways that regulate cell cycle progression and growth.
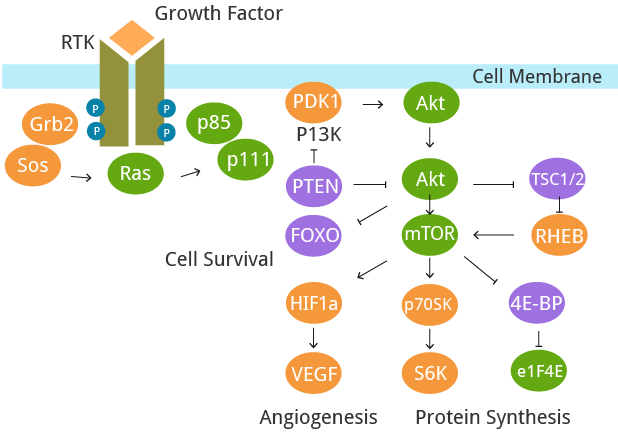
Fig. 2. The deregulation of signaling pathways.
Alternative Proliferative Signaling
Evading Growth Suppressors: Numerous tumor suppressors are involved in limiting cell growth and proliferation. They are often discovered through their characteristic inactivation in cancer cells. The two canonical tumor suppressors are the RB (retinoblastoma-associated) and TP53 proteins; they are essential regulators that govern apoptosis, proliferation, and senescence in cells.
Although TP53 and RB are essential regulators of cell-cycle progression, each operates as part of a more extensive network, allowing functional redundancy.
- In-house production of GF ligands and cognate receptors, leading to autocrine proliferative stimulation.
- Cancer cells may stimulate normal cells within the surrounding stroma, subsequently supplying the cancer cells with various growth factors.
- Increased GF receptor expression, rendering cells hyperresponsive.
- Structural alterations in the receptor molecules that increase ligand-independent activation.
- GF independence may arise from constitutive down-stream activation of signalling pathways, negating the need to stimulate these pathways via ligand-mediated receptor activation.
- The RB protein integrates signals from diverse extracellular and intracellular sources, acting as a critical gatekeeper of cell-cycle progression, its absence can result in persistent cell proliferation.
- TP53 receives input from intracellular stressors DNA damage, suboptimal growth conditions and homeostatic imbalances can lead to TP53 halting cell-cycle progression until conditions are favourable, or initiate apoptosis if cellular damage is beyond repair.
- The nuanced effects of activated TP53 are highly context dependent, varying by cell type as well as the severity of cellular stress and DNA damage.
Interestingly, chimeric mice populated with RB null cells do not demonstrate proliferative abnormalities- the only neoplasia observed were pituitary tumors later in life.1 TP53 null mice also develop typically, showing normal cellular and tissue homeostasis, but again develop cancers later in life, such as leukemia's and sarcomas.2
Activating Invasion and Metastasis: A well-characterized alteration to aid invasion and metastasis is the loss of E-cadherin by carcinoma cells. E-cadherin aids the assembly of epithelial cell sheets and maintains the cells' quiescence within these sheets by forming adherent junctions with adjacent epithelial cells. The frequently observed downregulation and occasional mutational inactivation of E-cadherin in human carcinomas provided strong support for its role as a viral suppressor of this hallmark capability.3 Another important driver of cancer metastasis is the loss of Contact inhibition. Contact inhibition ensures healthy, noncancerous cells cease proliferation and growth when they come into contact with each other. This characteristic is lost when cells undergo malignant transformation, leading to uncontrolled proliferation and solid tumor formation. 4
Contact Inhibition Mechanisms
- One mechanism involves the protein Merlin (coded by the NF2 gene), which strengthens cadherin-mediated cell-to-cell attachments.
- Sequestering growth factor receptors, Merlin limits the capability of cancer cells to efficiently emit mitogenic signals.
- The second mechanism of contact inhibition requires the LKB1 epithelial polarity protein, involved in tissue integrity.
- LKB1 has been shown to negate the mitogenic effects of the Myc oncogene when it is upregulated in organized, quiescent epithelial structures.
- When LKB1 expression is reduced, epithelial integrity is compromised, making epithelial cells more susceptible to Myc-induced transformation.
Expression of other adhesion molecules is notably altered in many carcinomas, with those involved in cytostasis being downregulated. Adhesion molecules generally associated with the cellular movement during embryogenesis and inflammation are predominantly upregulated. For example, N-cadherin, typically expressed in migrating neurons and mesenchymal cells during organogenesis, is upregulated in many invasive cancer cells.5
Enabling Replicative Immortality: Cancer cells require unlimited replicative potential to form macroscopic tumors, bypassing the Hayflick limit. Telomeres and telomerase play a crucial role in this hallmark of cancer.
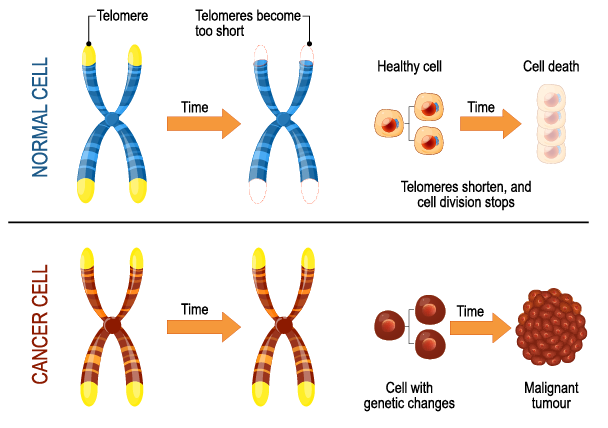
Fig. 3. Telomeres and Cancer.
- Telomere capping of chromosomes is heavily involved in cancer cells developing the capability for unlimited proliferation.
- Telomeres shorten progressively in non-immortalized cells, eventually losing the ability to protect the ends of chromosomes from end-to-end fusions.
- These fusions lead to the formation of dicentric chromosomes, which scramble the cell’s karyotype.
- Telomerase is predominantly absent in non-immortalized cells but expressed at functionally significant levels in the vast majority (̃90%) of spontaneously immortalized cells, such as human cancer cells.
- The presence of telomerase activity is correlated with a resistance to both senescence and apoptosis, which cancer cells must avoid to maintain replicative immortality.
Research suggests that cancer cells often experience telomere loss-induced crisis relatively early during multistep tumor progression due to their inability to express significant telomerase levels. Extensively eroded telomeres have been observed in premalignant growths, along with end-to-end chromosomal fusions, suggesting that cancer cells have passed through a substantial number of successive telomere-shortening cell divisions during their development from healthy cells before the acquisition of telomerase activity. 6
Inducing Angiogenesis: Cancer cells require nutrients and oxygen; tumor-associated vasculature formed through angiogenesis caters to these requirements. During tumor development, an "angiogenic switch" is almost always activated and remains on, causing normally quiescent vasculature to form new vessels.7
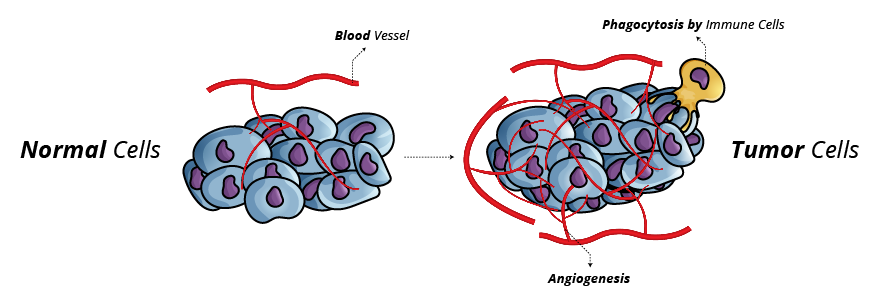
Fig. 4. Cancer Cell Angiogenesis
Angiogenesis Induction and Inhibition
The past decade has seen significant focus on angiogenesis. Amid this wealth of new knowledge, other proangiogenic signals, such as members of the fibroblast growth factor (FGF) family, have been implicated in sustaining tumor angiogenesis.8 Historically, angiogenesis was thought only to be relevant for the formation of rapidly growing tumors. However, recent investigation indicates that angiogenesis also plays a fundamental role in the premalignant phase of neoplastic progression.
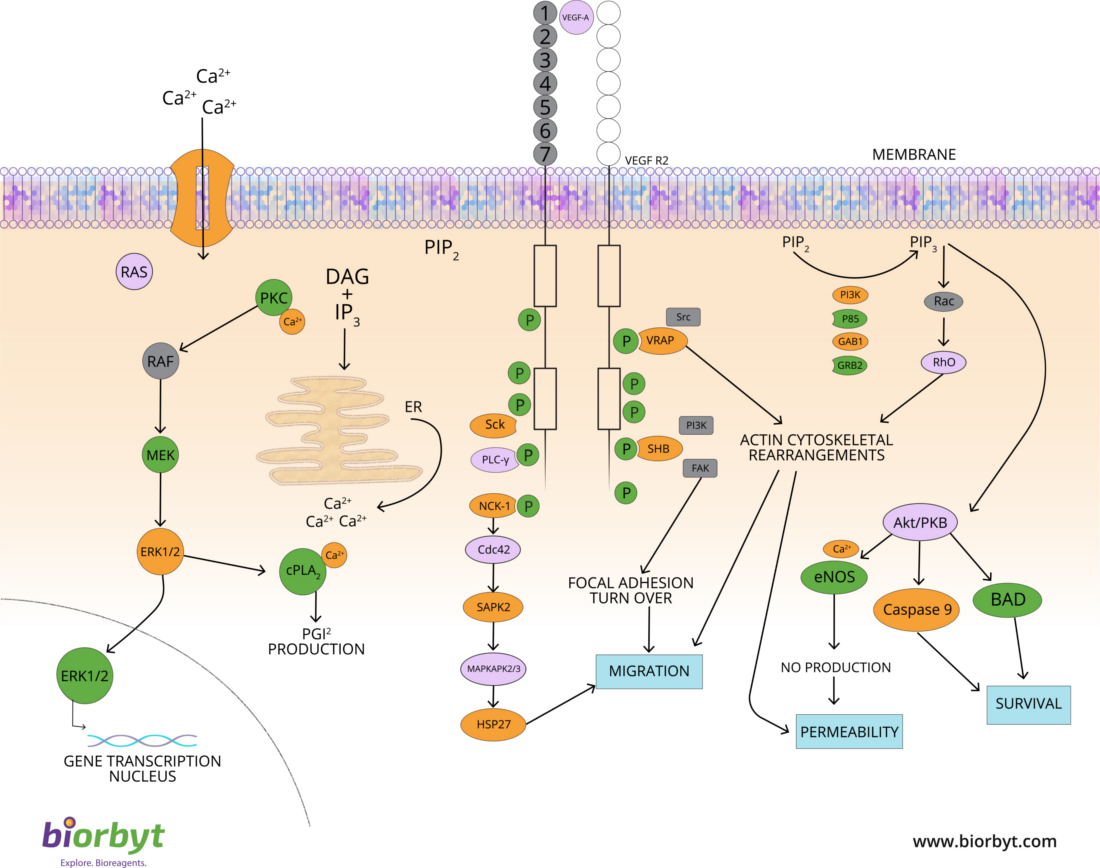
Fig. 5. VEGF Signaling Pathway
- VEGF signaling via the RTKs VEGFR-1–3 is regulated at multiple levels.
- VEGF gene expression is upregulated both by hypoxic conditions and via oncogenic signaling.
- TSP-1 binds transmembrane receptors displayed by endothelial cells, suppressing proangiogenic stimuli.
- The blood vessels produced within tumors are typically aberrant, displaying erratic capillary sprouting.
- Histological analyses of premalignant lesions indicate angiogenesis is induced surprisingly early during the multistage development of invasive cancers both in animal models and in humans.
- Once angiogenesis has been activated, tumours exhibit diverse patterns of neovascularization- pancreatic ductal adenocarcinomas are hypovascularized and may be actively antiangiogenic whereas human renal and pancreatic neuroendocrine carcinomas are densely vascularized.
Apoptosis
Apoptosis is the process of programmed cell death characterized by distinct morphological characteristics and biochemical mechanisms. It is a vital component of normal cell turnover, proper immune functioning, and embryonic development.
Resisting Cell Death: Programmed cell death via apoptosis serves as a genetic defence mechanism against cancer development. Regulators are subdivided into two primary circuits:
- The extrinsic program is involved in receiving and processing extracellular death-inducing signals.
- The intrinsic program, responsible for sensing and integrating intracellular signals.
Each program culminates in the activation of a predominantly latent protease (caspases 8 or 9), which proceeds to initiate a cascade of proteolysis involving effector caspases responsible for the execution phase of apoptosis, in which the cell is progressively disassembled and then consumed, both by its neighbors and by phagocytic cells. Currently, the intrinsic apoptotic program is more widely implicated as a barrier to cancer pathogenesis. 9
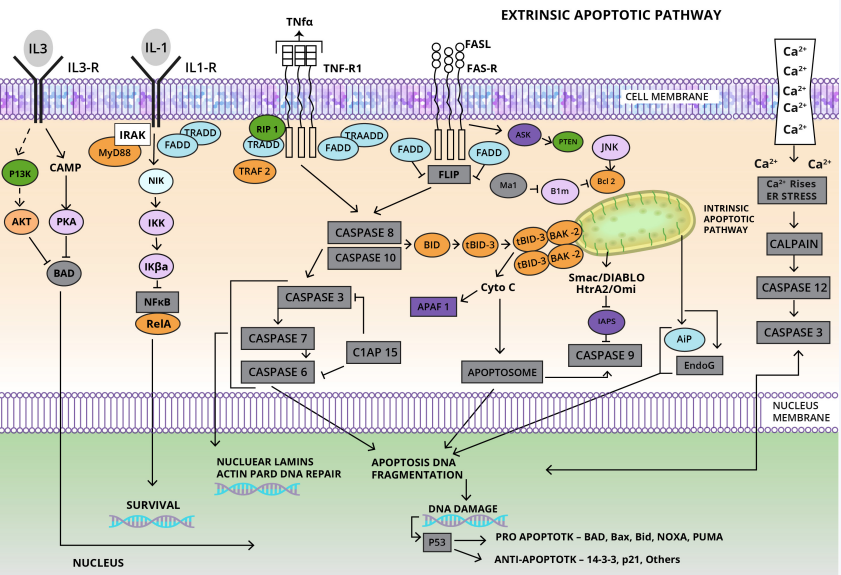
Fig. 6. Apoptosis Pathway
Apoptotic cells undergo morphological changes involving plasma membrane blebbing followed by karyorrhexis. Apoptotic bodies are subsequently phagocytosed by macrophages or neoplastic cells. Biochemical changes include the activation of caspases, breakdown of DNA, and membrane changes and recognition by phagocytic cells.
Evolved Strategies to Circumvent Apoptosis
- The most common mechanism involves losing the TP53 tumor suppressor function.
- Cancer cells may increase anti-apoptotic regulator expression, such as Bcl-2 or Bcl-xL.
- Alternatively, tumors can short-circuit the extrinsic death pathway.
Necrotic cell death releases proinflammatory signals into the microenvironment, which can recruit inflammatory cells that foster angiogenesis, proliferation, and invasiveness.13
The variety of apoptosis-avoiding mechanisms presumably reflects the diversity of apoptosis-inducing signals that cancer cell populations encounter during their evolution to the malignant state. In addition to apoptosis, necrosis also plays a role in resisting cell death; cell death by necrosis appears to be under genetic control in some situations, rather than being a random and uncoordinated process.1112
Necrotic cell death releases proinflammatory signals into the surrounding tissue microenvironment, allowing necrotic cells to recruit inflammatory cells. Evidence suggests that immune-inflammatory cells can be actively tumor-promoting in the context of cancer, as these cells can foster angiogenesis, cancer cell proliferation, and invasiveness. Furthermore, necrotic cells can release bioactive regulatory factors, such as IL-1α, which can directly stimulate neighboring viable cells to proliferate, with the potential to facilitate cancer progression.13
Emerging Hallmarks and Characteristics
Progress in the last decade has presented two further hallmarks: the reprogramming of energy metabolism and evading immune destruction. Two pre-requisite characteristics—genomic instability and tumor-promoting inflammation—enable these traits.
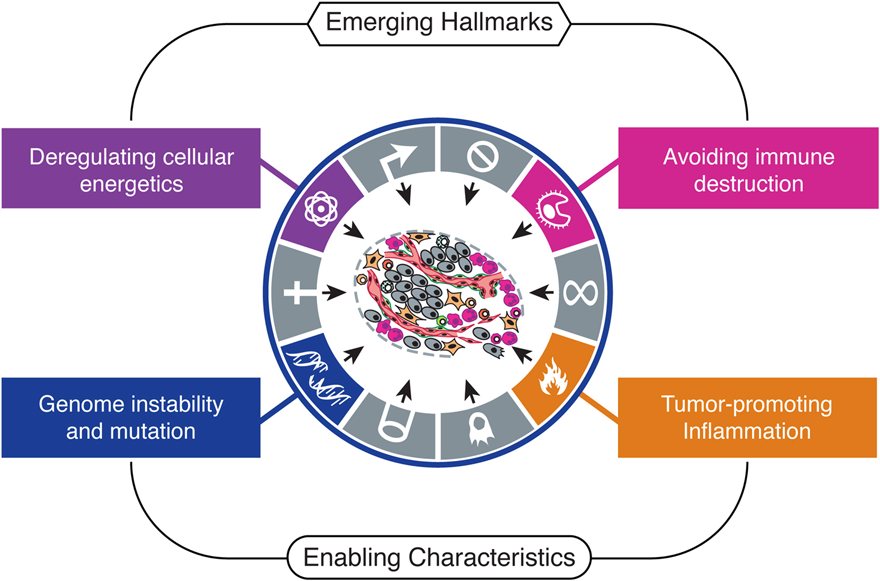
Fig. 7. Emerging Hallmarks and Enabling Characteristics. Source: Hanahan and Weinberg, 2011
Reprogramming Energy Metabolism
The chronic and predominantly unimpeded cell proliferation characteristic of cancer requires both the deregulated control of cell proliferation and energy metabolism adjustments, providing the fuel that enables cell growth and division. Cancer cell metabolism shows unique characteristics compared to normal cells; even in the presence of oxygen, cancer cells can alter their glucose metabolism and regulate their energy production. Cancer cells limit their energy metabolism primarily to glycolysis, resulting in a state coined aerobic glycolysis14 or the Warburg effect15. This reprogramming is achieved by upregulating glucose transporters, notably GLUT1, which substantially increases glucose import into the cytoplasm. 16
Such reprogramming of energy metabolism may appear counterintuitive, considering that cancer cells must compensate for an 18-fold lower efficiency of ATP production provided by glycolysis relative to normal mitochondrial oxidative phosphorylation. A functional rationale for the glycolytic switch in cancer cells has yet to be found. One hypothesis suggests that increased glycolysis allows the diversion of glycolytic intermediates into various biosynthetic pathways, including those generating nucleosides and amino acids. This, in turn, facilities the biosynthesis of the macromolecules and organelles necessary for assembling new cells. Furthermore, Warburg-like metabolism appears to be present in rapidly dividing embryonic tissues, suggesting a role in supporting the large-scale biosynthetic programs required for active cell proliferation seen in cancer.
Interestingly, some tumors have been found to contain two subpopulations of cancer cells that differ in their energy metabolism pathways. One population consists of aerobic glycolysis-dependant cells that secrete lactate. The second subpopulation preferentially imports and utilizes the lactate produced via the altered glycolytic pathway by neighboring cells their primary energy source, converting the exogenous lactate into pyruvate via Lactate dehydrogenases, which is then shuttled into the TCA cycle. 17
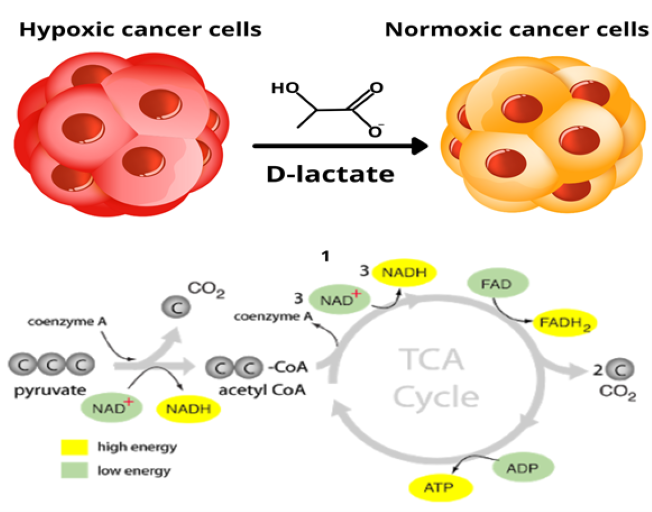
Fig. 8. Two subpopulations of cancer cells.
These two populations function symbiotically: the hypoxic cancer cells rely on glucose for fuel and secrete lactate as waste imported and preferentially used as fuel by their normoxic counterparts18. Furthermore, it has been shown that lactate stimulates angiogenesis through activation of the VEGF/VEGFR2 pathway, which may support the metastatic process 19. Altered energy metabolism is proving to be as widespread in cancer cells as many other cancer-associated traits that have been accepted as hallmarks of cancer.
Evading Immune Destruction
The long-standing theory of immune surveillance proposes that cells and tissues are constantly monitored by an ever-alert immune system which is responsible for recognizing and eliminating cancer cells, preventing tumor development; thus, for tumorigenesis to occur, cancerous cells must avoid detection by the immune system or limit the extent of immunological induced death, thereby evading eradication.
Previous research into carcinogen-induced cancers in genetically engineered immunodeficient mice observed that tumors arose more frequently and grew more rapidly when compared to immunocompetent controls. Functional deficiencies in CD8+ cytotoxic T lymphocytes (CTLs), CD4+ Th1 helper T cells, or natural killer (NK) cells each led to increases in tumor incidence, particularly in mice with combined immunodeficiencies in T cells and NK cells. The results indicated that both the innate and adaptive immune systems contribute significantly to immune surveillance and tumor eradication 20. Clinical evidence in cancer patients also suggests antitumoral immune responses in some forms of human cancer 21. Patients with colon and ovarian tumors heavily infiltrated with CTLs and NK cells show a better prognosis than those with reduced circulating killer lymphocytes22. Furthermore, some immunosuppressed organ transplant recipients have developed donor-derived cancers, indicating that in the tumor-free donors, the cancer cells were kept in a dormant state by a fully functional immune system 23.
Another hypothesis suggests highly immunogenic cancer cells may evade immune destruction by disabling the immune system's components responsible for eliminating them. Cancerous cells may inactivate infiltrating CTLs and NK cells through the secretion of TGF-β or other immunosuppressive factors24, 25 . More subtle mechanisms operate by recruiting inflammatory cells that are actively immunosuppressive, including regulatory T cells and myeloid-derived suppressor cells; both can suppress cytotoxic lymphocytes, aid cancer cell survival, and evade destruction by the immune system 26, 27.
Genomic Instability & Inflammation
Cancer cells can regulate inflammatory mechanisms to promote their growth and survival. During a normal response by the immune system, immune cells carry out their designated task of engulfing and destroying foreign invaders. Within the tumor microenvironment, immune cells are corrupted by cancer cells. As a result, the usually anti-tumor immune cells are subverted into tumor-promoting immune cells that secrete pro-survival, pro-migration, and anti-detection factors, enabling tumor growth metastasis. Important molecules and signaling pathways in mediating the immune response to the tumor microenvironment include NF-κB, inflammasome signaling, tumor-infiltrating immune cell markers, and immune checkpoint signaling 28.
NF-κB signaling in cancer and immune cells within the tumor microenvironment has been implicated in the epithelial-to-mesenchymal transition (EMT) of cells, allowing the detachment and migration the tumor mass. Crosstalk between NF-κB signaling in immune-infiltrating cells and cancer cells establishes an environment that promotes tumor growth, invasion, and malignancy 29. Research has shown that tumors engineer microenvironments to evade immune surveillance and attack, particularly by modulating specific immune checkpoint pathways 30.
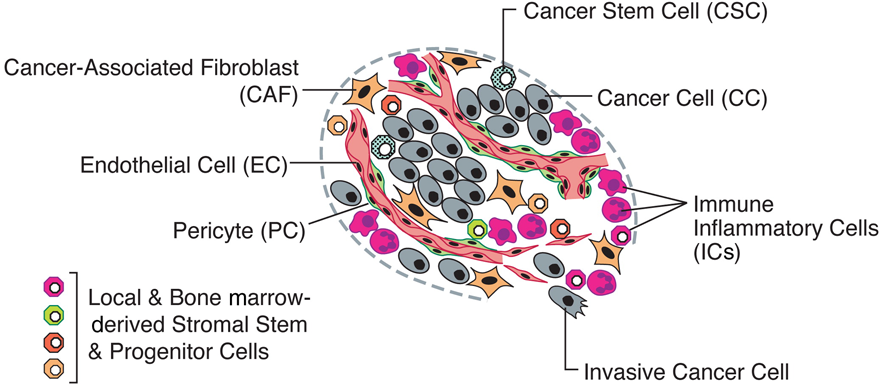
Fig. 9. An assemblage of distinct cell types constitutes most solid tumors.
T cells are the primary effector immune cells, expressing numerous autoinhibitory cell surface receptors, such as lymphocyte-activation gene 3 (LAG-3), programmed cell death protein 1 (PD-1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4); modulating cellular dynamics. Within the tumor microenvironment, tumor cells upregulate the ligands to these receptors, enhancing tumor tolerance to immunological assault, enabling evasion and subsequent eradication by the immune system 31.
Pharmacological modulators of these ligand-receptor interactions, known as immune checkpoint therapies, have been intensely researched and deployed as novel immunotherapy agents to treat cancers in recent years. Of particular interest are monoclonal antibodies against PD-1 and CTLA-4. Given these immune checkpoint therapies' early success in activating anti-tumor immune responses, creating immunotherapies targeting other co-inhibitory and co-stimulatory receptors and their ligands in an order appears to be a compelling therapeutic strategy 32.
instability is a well-known characteristic of most cancers. From sustaining proliferative signaling to avoiding cell death, genome instability generates the genetic abnormalities required for multiple hallmark functions. Either microsatellite instability or chromosomal abnormalities predominantly cause hereditary cancers; the basis for this underlying genomic instability is due to mutations in DNA repair genes. In contrast, The pattern of mutations in sporadic human cancers indicates that the selective pressure for tumor suppressor p53 (TP53) mutations is linked to DNA damage rather than p14ARF activation. Genomic instability in sporadic human cancers has also been linked to oncogene-induced DNA damage 33.
Conclusion
The hallmarks of cancer comprise of both well-established and numerous emerging biological capabilities acquired during the multistep development of human tumors. These hallmarks constitute an organizing principle for rationalizing the complexities of cancer development and progression. From sustaining proliferative signaling to avoiding cell death, genome instability generates the genetic abnormalities required for multiple hallmark functions. The Conceptual progress of the hallmarks of cancer in the last decade has presented two further hallmarks, the reprogramming of energy metabolism and evading immune destruction. In addition to cancer cells, tumors exhibit another dimension of complexity: they contain a repertoire of recruited, ostensibly normal cells that contribute to the acquisition of hallmark traits by creating the "tumor microenvironment." Recognition of the widespread applicability of these concepts will increasingly affect the development of new means to treat human cancer.
References
- Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene. 1999 Dec 20;18(55):7873-82. doi: 10.1038/sj.onc.1203244. PMID: 10630640.
- Ghebranious N, Donehower LA. Mouse models in tumor suppression. Oncogene. 1998 Dec 24;17(25):3385-400. doi: 10.1038/sj.onc.1202573. PMID: 9917000
- Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009 Dec;1(6):a003129. doi: 10.1101/cshperspect.a003129. Epub 2009 Sep 23. PMID: 20457567; PMCID: PMC2882122.
- Pavel, M., Renna, M., Park, S.J. et al. Contact inhibition controls cell survival and proliferation via YAP/TAZ-autophagy axis. Nat Commun 9, 2961 (2018). https://doi.org/10.1038/s41467-018-05388-x.
- Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004 Feb;4(2):118-32. doi: 10.1038/nrc1276. PMID: 14964308.
- Cleal K, Norris K, Baird D. Telomere Length Dynamics and the Evolution of Cancer Genome Architecture. Int J Mol Sci. 2018;19(2):482. Published 2018 Feb 6. doi:10.3390/ijms19020482.
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996 Aug 9;86(3):353-64. doi: 10.1016/s0092-8674(00)80108-7. PMID: 8756718.
- Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009 Oct;19(5):329-37. doi: 10.1016/j.semcancer.2009.05.003. Epub 2009 May 29. PMID: 19482086.
- Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007 Feb 26;26(9):1324-37. doi: 10.1038/sj.onc.1210220. PMID: 17322918; PMCID: PMC2930981.
- Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008 Dec 26;135(7):1161-3. doi: 10.1016/j.cell.2008.12.004. PMID: 19109884.
- Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006 Jan 1;20(1):1-15. doi: 10.1101/gad.1376506. PMID: 16391229.
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010 Mar 19;140(6):883-99. doi: 10.1016/j.cell.2010.01.025. PMID: 20303878; PMCID: PMC2866629.
- Peggy P. Hsu1,2 and David M. Sabatini1,2,3,* 1 Whitehead Institute for Biomedical Research and Massachusetts Institute of Technology Department of Biology, Cambridge, MA 02142, USA 2 Broad Institute, Cambridge, MA 02142, USA 3 Koch Institute for Integrative Cancer Research at MIT, Cambridge, MA 02139, USA *Correspondence: sabatini@wi.mit.edu DOI 10.1016/j.cell.2008.08.021.
- Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? [published correction appears in Trends Biochem Sci. 2016 Mar;41(3):287] [published correction appears in Trends Biochem Sci. 2016 Mar;41(3):287]. Trends Biochem Sci. 2016;41(3):211-218. doi:10.1016/j.tibs.2015.12.001.
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009 Mar 1;23(5):537-48. doi: 10.1101/gad.1756509. PMID: 19270154; PMCID: PMC2763495.
- Kennedy KM, Dewhirst MW. Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. 2010 Jan;6(1):127-48. doi: 10.2217/fon.09.145. PMID: 20021214; PMCID: PMC2819205.
- San-Millán I, Brooks GA. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis. 2017;38(2):119-133. doi:10.1093/carcin/bgw127.
- Dhup S, Dadhich RK, Porporato PE, Sonveaux P. Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr Pharm Des. 2012;18(10):1319-30. doi: 10.2174/138161212799504902. PMID: 22360558.
- Teng MW, Swann JB, Koebel CM, Schreiber RD, Smyth MJ. Immune-mediated dormancy: an equilibrium with cancer. J Leukoc Biol. 2008 Oct;84(4):988-93. doi: 10.1189/jlb.1107774. Epub 2008 May 30. PMID: 18515327.
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, Bruneval P, Fridman WH, Becker C, Pagès F, Speicher MR, Trajanoski Z, Galon J. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013 Oct 17;39(4):782-95. doi: 10.1016/j.immuni.2013.10.003. PMID: 24138885.
- Pagès F, Galon J, Dieu-Nosjean MC, Tartour E, Sautès-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010 Feb 25;29(8):1093-102. doi: 10.1038/onc.2009.416. Epub 2009 Nov 30. PMID: 19946335.
- Strauss DC, Thomas JM. Transmission of donor melanoma by organ transplantation. Lancet Oncol. 2010 Aug;11(8):790-6. doi: 10.1016/S1470-2045(10)70024-3. Epub 2010 May 5. PMID: 20451456.
- Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010 May 7;328(5979):749-52. doi: 10.1126/science.1185837. Epub 2010 Mar 25. PMID: 20339029.
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008 Jun;14(6):818-29. doi: 10.1016/j.devcel.2008.05.009. PMID: 18539112.
- Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res. 2010;107:57-117. doi: 10.1016/S0065-230X(10)07003-X. PMID: 20399961.
- Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009 Apr 15;182(8):4499-506. doi: 10.4049/jimmunol.0802740. PMID: 19342621; PMCID: PMC2810498.
- Grivennikov SI, Karin M. Inflammation and oncogenesis: A vicious connection. Curr Opin Genet Dev 2010;20:65-71.
- Fares, J., Fares, M.Y., Khachfe, H.H. et al. Molecular principles of metastasis: a hallmark of cancer revisited. Sig Transduct Target Ther 5, 28 (2020). https://doi.org/10.1038/s41392-020-0134-x.
- Li X, Shao C, Shi Y, Han W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J Hematol Oncol. 2018;11(1):31. Published 2018 Feb 27. doi:10.1186/s13045-018-0578-4.
- Chae YK, Arya A, Iams W, et al Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC)Journal for ImmunoTherapy of Cancer 2018;6:39. doi: 10.1186/s40425-018-0349-3.
- Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol. 2018;8:86. Published 2018 Mar 28. doi:10.3389/fonc.2018.00086.
- Negrini, S., Gorgoulis, V. & Halazonetis, T. Genomic instability — an evolving hallmark of cancer. Nat Rev Mol Cell Biol 11, 220–228 (2010). https://doi.org/10.1038/nrm2858.
Sources
- International Agency for Research on Cancer
- Cancer Research UK
- Hallmarks of Cancer- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74. doi: 10.1016/j.cell.2011.02.013. PMID: 21376230
- Innovative Approaches for Cancer Treatment- Pucci C, Martinelli C, Ciofani G. Innovative approaches for cancer treatment: current perspectives and new challenges. Ecancermedicalscience. 2019;13:961. doi: 10.3332/ecancer.2019.961. PMID: 31537986; PMCID: PMC6753017.
- Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. BY MATTHEW G. VANDER HEIDEN, LEWIS C. CANTLEY, CRAIG B. THOMPSON SCIENCE22 MAY 2009: 1029-1033
- Bald, T., Krummel, M.F., Smyth, M.J. et al. The NK cell–cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nat Immunol 21, 835–847 (2020). https://doi.org/10.1038/s41590-020-0728-z